
Chapter 13 Single-cell RNA-seq
This chapter is in a beta stage. If you wish to contribute, please go to this form or our GitHub page.

13.2 Where single-cell RNA-seq data comes from
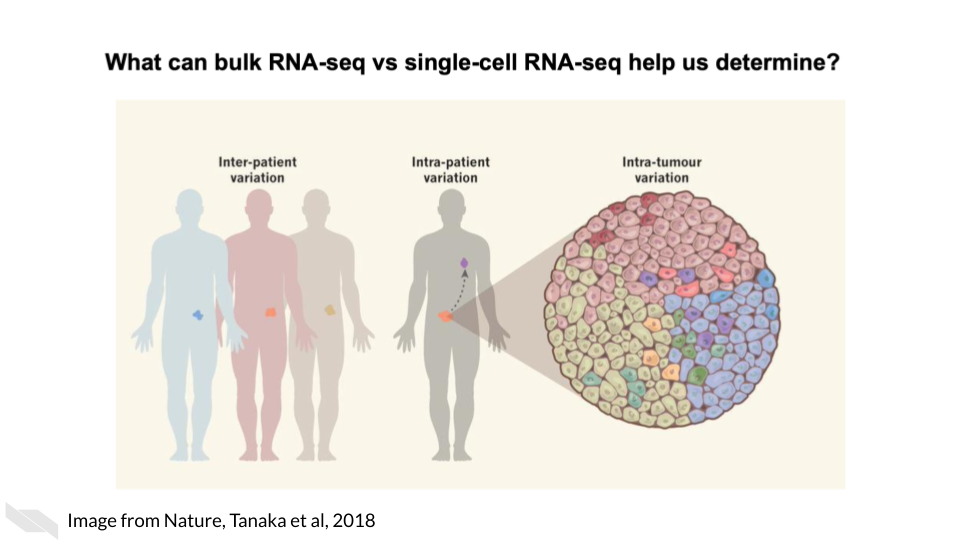
As opposed to bulk RNA-seq which can only tell us about tissue level and within patient variation, single-cell RNA-seq is able to tell us cell to cell variation in transcriptomics including intra-tumor heterogeneity.
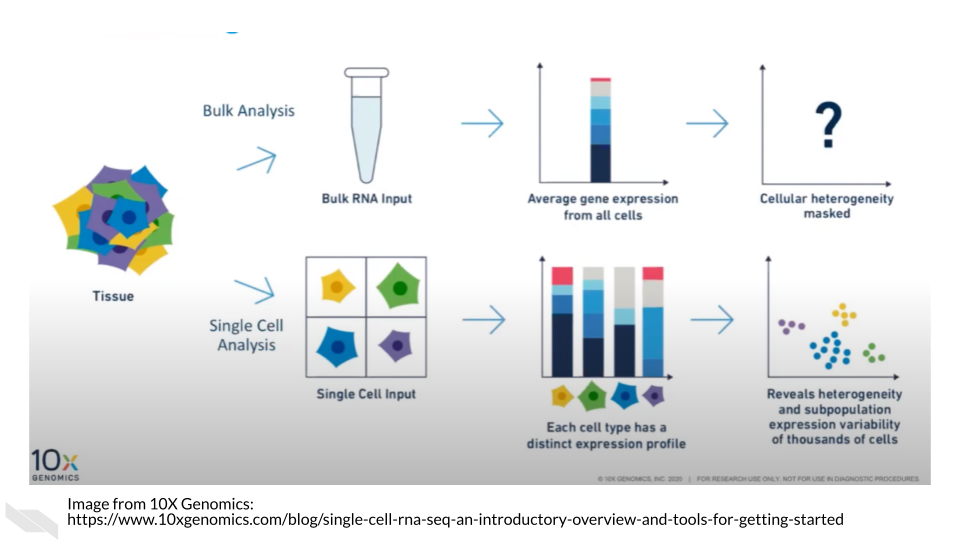
Single cell RNA-seq can give us cell level transcriptional profiles. Whereas bulk RNA-seq masks cell to cell heterogeneity. If your research questions require cell-level transcriptional information, single-cell RNA-seq will on interest to you.
13.3 Single-cell RNA-seq data types
There are broadly two categories of single-cell RNA-seq data methods we will discuss.
- Full length RNA-seq: Individual cells are physically separated and then sequenced.
- Tag Based RNA-seq: Individual cells are tagged with a barcode and their data is separated computationally.
Depending on your goals for your single cell RNA-seq analysis, you may want to choose one method over the other.


(Material borrowed from (“Alex’s Lemonade Training Modules” 2022)).
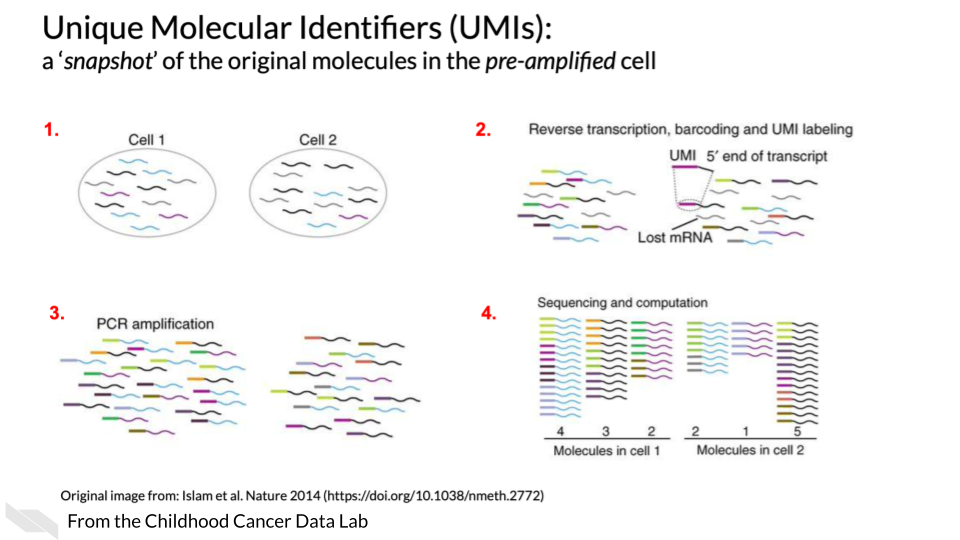
13.3.1 Unique Molecular identifiers
Often Tag based single cell RNA-seq methods will include not only a cell barcode for cell identification but will also have a unique molecular identifier (UMI) for original molecule identification. The idea behind the UMIs is it is a way to have insight into the original snapshot of the cell and potentially combat PCR amplification biases.
13.4 Single cell RNA-seq tools
There are a lot of scRNA-seq tools for various steps along the way.
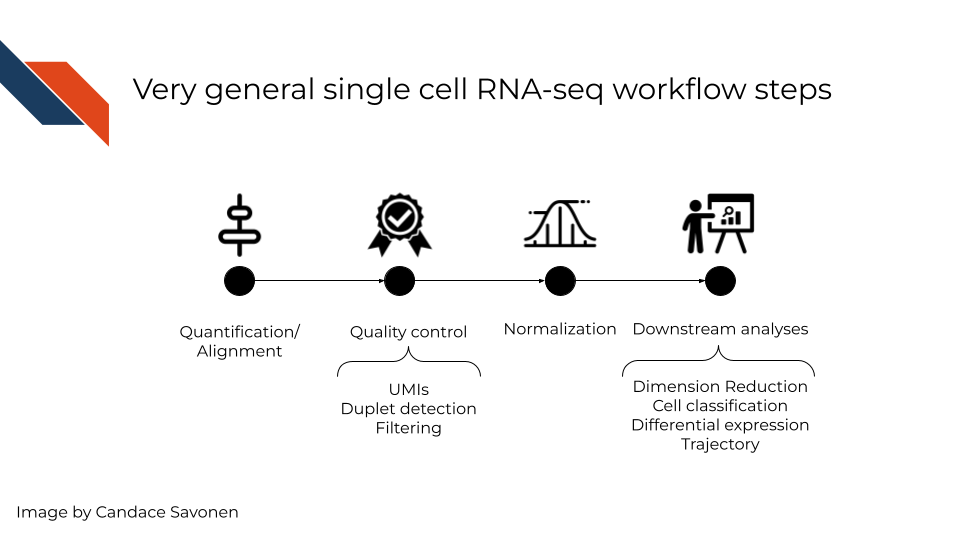
In a very general sense, single cell RNA-seq workflows involves first quantification/alignment. You will also need to conduct quality control steps that may involve using UMIs to check for what’s detected, detecting doublets (also known as duplets), and using this information to filter out data that is not trustworthy. Doublets are transcriptome data generated from two cells, and an undesired technical artifact when single cell RNA-seq workflows want data representing a single cell at a time. After you have a set of reliable data, you need to normalize your data. Single cell data is highly skewed - a lot of genes barely or not detected and a few genes that are detected a lot. After data has been normalized you are ready to conduct your downstream analyses. This will be highly dependent on the original goals and questions of your experiment. It may include dimension reduction, cell classification, differential expression, detecting cell trajectories or any number of other analyses.
Each step of this very general representation of a workflow can be conducted by a variety of tools. We will highlight some of the more popular tools here. But, to look through a full list, you can consult the scRNA-tools website.
13.5 Quantification and alignment tools
This following pros and cons sections have been written by AI (Claude 3.7 Sonnet) and may need verification by experts. This is meant to give you a basic idea of the pros and cons of these tools but should ultimately be used with your own judgment.
- STAR (Dobin et al. 2013):
- Pros: Accurate alignment of RNA-seq reads to the genome. Can handle a wide range of RNA-seq protocols, including scRNA-seq. Provides read counts and gene-level expression values.
- Cons: Requires a significant amount of memory and computational resources. May be difficult to set up and run for beginners.
- HISAT2 (Kim, Langmead, and Salzberg 2015):
- Pros: Accurate alignment of RNA-seq reads to the genome. Provides transcript-level expression values. Supports splice-aware alignment.
- Cons: May require significant computational resources for large datasets. May not be as accurate as some other alignment tools.
- Kallisto bustools (Bray et al. 2016):
- Pros: Fast and accurate quantification of RNA-seq reads without the need for alignment. Provides transcript-level expression values. Requires less memory and computational resources than alignment-based methods.
- Cons: May not be as accurate as alignment-based methods for lowly expressed genes. Cannot provide allele-specific expression estimates.
- Alevin/Salmon (Patro et al. 2017):
- Pros: Fast and accurate quantification of RNA-seq reads without the need for alignment. Provides transcript-level expression values. Supports both single-end and paired-end sequencing.
- Cons: May not be as accurate as alignment-based methods for lowly expressed genes. Cannot provide allele-specific expression estimates.
- Cell Ranger (Zheng et al. 2017):
- Pros: Specifically designed for 10x Genomics scRNA-seq data, with optimized workflows for alignment and quantification. Provides read counts and gene-level expression values. Offers a streamlined pipeline with minimal input from the user.
- Cons: Limited options for customizing parameters or analysis methods. May not be suitable for datasets from other scRNA-seq platforms.
13.6 Downstream tools Pros and Cons
- Seurat:
- Pros: Has a wide range of functionalities for preprocessing, clustering, differential expression, and visualization. Can handle multiple modalities, including CITE-seq and ATAC-seq. Has a large and active user community, with extensive documentation and tutorials available.
- Cons: Can be computationally intensive, especially for large datasets. Requires some knowledge of R programming language.
- Scanpy:
- Pros: Written in Python, a widely used programming language in bioinformatics. Has a user-friendly interface and extensive documentation. Offers a variety of preprocessing, clustering, and differential expression methods, as well as interactive visualizations.
- Cons: May not be as feature-rich as some other tools, such as Seurat. Does not yet support multiple modalities.
- Monocle:
- Pros:Focuses on trajectory analysis, allowing users to explore developmental trajectories and cell fate decisions. Has a user-friendly interface and extensive documentation. Can handle data from multiple platforms, including Smart-seq2 and Drop-seq.
- Cons: May not be as feature-rich for clustering or differential expression analysis as some other tools. Requires some knowledge of R programming language.
- Monocle:
- Pros:Focuses on trajectory analysis, allowing users to explore developmental trajectories and cell fate decisions. Has a user-friendly interface and extensive documentation. Can handle data from multiple platforms, including Smart-seq2 and Drop-seq.
- Cons: May not be as feature-rich for clustering or differential expression analysis as some other tools. Requires some knowledge of R programming language.
13.6.1 Doublet Tool Pros and Cons
- DoubletFinder(McGinnis, Murrow, and Gartner 2020):
- Pros: Uses a machine learning approach to detect doublets based on transcriptome similarity. Can be used with a variety of scRNA-seq platforms. Offers a user-friendly interface and extensive documentation.
- Cons: Can be computationally intensive for large datasets. May require some knowledge of R programming language.
- Scrublet (Wolock, Krishnaswamy, and Huang 2019):
- Pros: Uses a density-based approach to detect doublets based on barcode sharing. Fast and computationally efficient, making it suitable for large datasets. Offers a user-friendly interface and extensive documentation.
- Cons:May not be as accurate as other methods, especially for low-quality data. Limited to 10x Genomics data.
- DoubletDecon (De Pasquale and Dudoit 2019):
- Pros: Uses a statistical approach to identify doublets based on the distribution of the number of unique molecular identifiers (UMIs) per cell. Can be used with different platforms and species. Offers a user-friendly interface and extensive documentation.
- Cons: May not be as accurate as other methods, especially for data with low sequencing depth or low cell numbers. Requires some knowledge of R programming language.
It’s important to note that no doublet detection method is perfect, and it’s often a good idea to combine multiple methods to increase the accuracy of doublet identification. Additionally, manual inspection of the data is always recommended to confirm the presence or absence of doublets.
13.7 More scRNA-seq tools and tutorials
- AlevinQC
- Gene Pattern’s single cell RNA-seq tutorials - an open software environment providing access to hundreds of tools for the analysis and visualization of genomic data.
- Single Cell Genome Viewer
- For normalization scater
- TumorDecon can be used to generate customized signature matrices from single-cell RNA-sequence profiles. It is available on Github (https://github.com/ShahriyariLab/TumorDecon) and PyPI (https://pypi.org/project/TumorDecon/).
13.8 Visualization GUI tools
- WebMeV uniquely provides a user-friendly, intuitive, interactive interface to processed analytical data uses cloud-computing elasticity for computationally intensive analyses and is compatible with single cell or bulk RNA-seq input data.
- UCSC Xena is a web-based visualization tool for multi-omic data and associated clinical and phenotypic annotations. It can be used with single cell RNA-seq data.
- Integrative Genomics Viewer (IGV) is a track-based browser for interactively exploring genomic data mapped to a reference genome.
13.9 Useful tutorials
These tutorials cover explicit steps, code, tool recommendations and other considerations for analyzing RNA-seq data.
- Orchestrating Single Cell Analysis with Bioconductor - An excellent tutorial for processing single cell data using Bioconductor.
- Advanced Single Cell Analysis with Bioconductor - a companion book to the intro version that contains code examples.
- Alex’s Lemonade scRNA-seq Training module - A cancer based workshop module based in R, with exercise notebooks.
- Sanger Single Cell Course - a general tutorial based on using R.
- ASAP: Automated Single-cell Analysis Pipeline is a web server that allows you to process scRNA-seq data.
- Processing raw 10X Genomics single-cell RNA-seq data (with cellranger) - a tutorial based on using CellRanger.
13.10 Useful readings
- An Introduction to the Analysis of Single-Cell RNA-Sequencing Data (AlJanahi, Danielsen, and Dunbar 2018).
- Orchestrating single-cell analysis with Bioconductor (Amezquita et al. 2020).
- UMIs the problem, the solution and the proof (Smith 2015).
- Experimental design for single-cell RNA sequencing (Baran-Gale, Chandra, and Kirschner 2018).
- Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies (Lafzi et al. 2018).
- Comparative Analysis of Single-Cell RNA Sequencing Methods (Ziegenhain et al. 2017).
- Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems (X. Zhang et al. 2019).
- Single cells make big data: New challenges and opportunities in transcriptomics (Angerer et al. 2017).
- Comparative Analysis of common alignment tools for single cell RNA sequencing (Brüning et al. 2021).
- Current best practices in single-cell RNA-seq analysis: a tutorial (Luecken and Theis 2019).