
Chapter 15 Chromatin Methods Overview
This chapter is incomplete! If you wish to contribute, please go to this form or our GitHub page.
In its existing form, this chapter has been written with AI (Claude 3.7 Sonnet) and still needs further verification by experts.

15.2 Why are people interested in chromatin?
Chromatin plays a crucial role in regulating gene expression, which is essential for a wide range of biological processes. It is the complex of DNA and proteins that make up the structure of chromosomes in the nucleus of a cell. The DNA in chromatin is packaged around histone proteins in a way that can either promote or inhibit access to the DNA by other proteins that control gene expression. Specifically, chromatin structure can affect the ability of transcription factors and RNA polymerase to bind to and transcribe genes.
Changes in chromatin structure can lead to changes in gene expression, which can have profound effects on cell function and development. For example, chromatin remodeling is a key step in cell differentiation, during which cells become specialized and take on specific functions. Dysregulation of chromatin structure can also lead to the development of diseases, such as cancer, in which aberrant gene expression contributes to uncontrolled cell growth and proliferation.
Therefore, understanding the mechanisms that regulate chromatin structure and function is crucial for advancing our understanding of cellular processes, disease development, and potential therapies. This is why chromatin research has become a major area of focus in molecular biology and genomics research.
15.3 What kinds of questions can chromatin answer?
- How are genes turned on and off in response to developmental cues or environmental stimuli?
- What are the mechanisms by which chromatin structure is altered during cell differentiation and development?
- How do epigenetic modifications, such as DNA methylation and histone modifications, affect chromatin structure and gene expression?
- How does chromatin structure influence the binding of transcription factors and other regulatory proteins to specific regions of the genome?
- How is chromatin structure altered in diseases such as cancer, and how can this knowledge be used to develop new therapies?
- How can we manipulate chromatin structure to selectively activate or repress specific genes, and what are the potential applications of such approaches?
15.3.1 Chromatin is involved in a variety of biological processes:
- Gene expression: Chromatin structure and organization play a crucial role in regulating gene expression. The packaging of DNA around histone proteins can either promote or inhibit access to the DNA by other proteins that control gene expression.
- DNA replication and repair: Chromatin structure can also affect DNA replication and repair. For example, histone modifications and chromatin remodeling can facilitate access to DNA replication and repair machinery.
- Epigenetic regulation: Epigenetic modifications, such as DNA methylation and histone modifications, can be stably inherited and play a critical role in the regulation of gene expression.
- Cell differentiation: Chromatin structure is dynamically regulated during cell differentiation and plays a key role in determining cell fate and function.
- Development: Chromatin structure also plays an important role in the regulation of developmental processes, such as morphogenesis and organogenesis.
- Disease: Dysregulation of chromatin structure and function is associated with a wide range of diseases, including cancer, neurodegenerative disorders, and developmental disorders.
15.4 Comparison of technologies
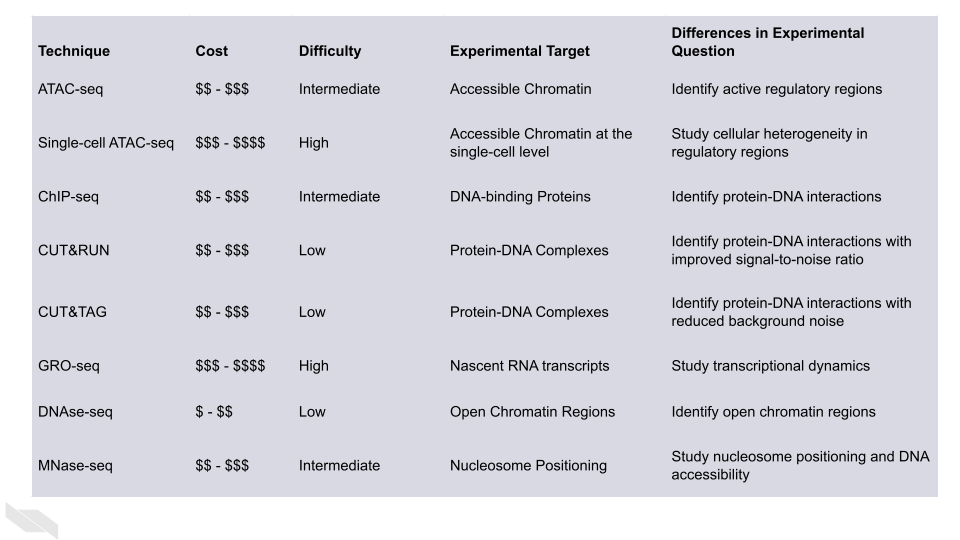
15.4.1 ATAC-seq:
ATAC-seq (Assay for Transposase Accessible Chromatin using sequencing) is a technique that uses transposases to fragment DNA and insert sequencing adapters into accessible chromatin regions. The DNA fragments are then sequenced to identify regions of open chromatin. This technique is widely used to study the epigenetic regulation of gene expression.
15.4.1.1 When to use ATAC-seq:
- When you want to study the epigenetic regulation of gene expression.
- When you want to identify open chromatin regions associated with regulatory elements such as enhancers and promoters.
- When you want to study various cell types and tissues, including difficult-to-access cell types.
15.4.1.2 Advantages:
- ATAC-seq is a simple and cost-effective technique that requires a low amount of starting material.
- It allows the identification of open chromatin regions, which are usually associated with regulatory elements such as enhancers and promoters.
- ATAC-seq can be used to study various cell types and tissues, including difficult-to-access cell types.
15.4.1.3 Disadvantages:
- ATAC-seq can have high background noise due to non-specific cleavage of chromatin.
- It may miss lowly accessible regions due to a bias towards highly accessible regions.
- It is difficult to identify the specific regulatory elements that are associated with open chromatin regions.
15.4.2 Single-cell ATAC-seq:
Single-cell ATAC-seq is a technique that combines single-cell sequencing and ATAC-seq to identify open chromatin regions in individual cells. This technique allows the study of epigenetic heterogeneity between cells and the identification of cell-specific regulatory elements.
15.4.2.1 When to use single-cell ATAC-seq:
- When you want to study the epigenetic heterogeneity between cells and identify cell-specific regulatory elements.
- When you want to identify rare cell types or rare cell states that may be missed by bulk techniques.
- When you want to study the epigenetic dynamics of cells in response to environmental changes.
15.4.2.2 Advantages:
- Single-cell ATAC-seq allows the identification of open chromatin regions in individual cells, which provides cell-specific epigenetic information.
- It can identify rare cell types and rare cell states that may be missed by bulk techniques.
- It can be used to study the epigenetic dynamics of cells in response to environmental changes.
15.4.2.3 Disadvantages:
- Single-cell ATAC-seq can have a higher level of technical noise due to the low amount of starting material.
- It can be challenging to obtain high-quality single-cell suspensions from tissues.
- It can be difficult to analyze the large amount of data generated by single-cell sequencing techniques.
15.4.3 ChIP-seq:
ChIP-seq (Chromatin Immunoprecipitation sequencing) is a technique that uses antibodies to isolate specific DNA-protein complexes, such as transcription factors or histone modifications. The DNA fragments associated with the protein complexes are then sequenced to identify the genomic regions that are bound by the protein.
15.4.3.1 Advantages:
- ChIP-seq allows the identification of specific protein-DNA interactions, which provides information on the regulation of gene expression.
- It can be used to study the epigenetic changes associated with specific cellular processes, such as differentiation or development.
- ChIP-seq can identify the binding sites of transcription factors, which can be used to identify regulatory elements such as enhancers and promoters.
15.4.4 CUT&RUN
CUT&RUN (Cleavage Under Targets & Release Using Nuclease) is a relatively new genomic method that involves the targeted cleavage of DNA by a specific antibody or protein of interest, followed by the release and sequencing of the DNA fragments. The CUT&RUN method was developed as a more streamlined alternative to the ChIP-seq (Chromatin Immunoprecipitation sequencing) method, which involves a more complex series of steps Skene and Henikoff (2018).
15.4.4.1 How CUT&RUN works:
Cells are permeabilized and incubated with a specific antibody or protein of interest. This antibody or protein is fused to a protein called Protein A-Micrococcal Nuclease (pA-MNase). After incubation, the pA-MNase is activated and cleaves the DNA in the vicinity of the bound antibody or protein of interest. The released DNA fragments are then purified and sequenced to identify the genomic regions that were bound by the antibody or protein of interest.
CUT&RUN has several advantages over ChIP-seq, including:
- CUT&RUN requires a lower amount of starting material and can be performed more quickly than ChIP-seq.
- CUT&RUN produces less background noise, as the DNA is cleaved in situ, rather than being fragmented by sonication or other methods.
- CUT&RUN can be used to study chromatin-associated proteins that may not be easily solubilized for ChIP-seq.
15.4.5 CUT&Tag
CUT&Tag (Cleavage Under Targets and Tagmentation) is similar to CUT&RUN. It was developed as an improvement over CUT&RUN, with the goal of reducing the amount of background noise and improving the efficiency of the method (Kaya-Okur et al. 2019).
15.4.5.1 How CUT&Tag works:
- Cells are permeabilized and incubated with a specific antibody or protein of interest, which is fused to a protein called Protein A-Tn5 transposase.
- The Protein A-Tn5 transposase inserts sequencing adapters into the genomic DNA in the vicinity of the bound antibody or protein of interest.
- The DNA is then released from the chromatin by the Protein A-Tn5 transposase and purified for sequencing.
Like CUT&RUN, CUT&Tag allows for the specific cleavage of DNA in the vicinity of a target protein or antibody, but the addition of sequencing adapters in CUT&Tag occurs directly in the nucleus, prior to DNA release. This results in less background noise and more efficient DNA recovery.
15.4.5.2 Advantages:
- CUT&Tag has a lower level of background noise and higher sensitivity due to the addition of sequencing adapters in situ.
- CUT&Tag requires less input material than CUT&RUN, which makes it a more efficient method.
- CUT&Tag can be used to study the binding sites of transcription factors and chromatin-associated proteins.
Overall, both CUT&RUN and CUT&Tag are powerful genomic methods that allow for the efficient study of protein-DNA interactions and epigenetics. The choice between the two methods may depend on the specific research question and the availability of specific reagents or equipment.
15.4.6 GRO-seq (Global Run-On sequencing)
Allows for the genome-wide analysis of transcriptional activity by measuring the nascent RNA transcripts that are actively being synthesized by RNA polymerase. GRO-seq is a high-throughput sequencing-based technique that provides a snapshot of the transcriptional landscape of a cell Park and Won (2018).
15.4.7 How GRO-seq works:
- Nuclei are isolated from cells and incubated with a biotinylated nucleotide triphosphate, which is incorporated into nascent RNA transcripts by RNA polymerase.
- The labeled RNA is then selectively captured using streptavidin beads, and the RNA is reverse-transcribed into cDNA.
- The cDNA is then sequenced to identify the regions of the genome that are actively transcribed.
15.4.7.1 Advantages:
- Its ability to distinguish between the sense and antisense strands of transcribed RNA
- Its ability to quantify the level of transcriptional activity in individual genes
- Its ability to identify novel transcripts and transcriptional start sites.
DNase-seq and MNase-seq are alternative approaches which can be used to identify accessible regions of chromatin. MNase-seq is particularly useful for studying the occupancy of nucleosomes or transcription factors with high resolution. DNase-seq uses DNAse I to cleave DNA at hypersensitive sites typically associated with cis-regulatory elements. It is also possible to footprint TF occupancy with base-pair level resolution using DNase-seq, while the quality of ATAC-seq footprinting is still in question. Additionally, although both DNAse-seq and MNase-seq have sequence biases as well, the sequence preference is different for each enzyme.