
Chapter 2 A Very General Genomics Overview
2.1 Learning Objectives

In this chapter we are going to cover sequencing and microarray workflows at a very general high level overview to give you a first orientation. As we dive into specific data types and experiments, we will get into more specifics. Here we will cover the most common file formats. If you have a file format you are dealing with that you don’t see listed here, it may be specific to your data type and we will discuss that more in that data type’s respective chapter. We still suggest you go through this chapter to give you a basic understanding of commonalities of all genomic data types and workflows
2.1.1 What do genomics workflows look like?
In the most general sense, all genomics data when originally collected is raw, it needs to undergo processing to be normalized and ready to use. Then normalized data is generally summarized in a way that is ready for it to be further consumed. Lastly, this summarized data is what can be used to make inferences and create plots and results tables.
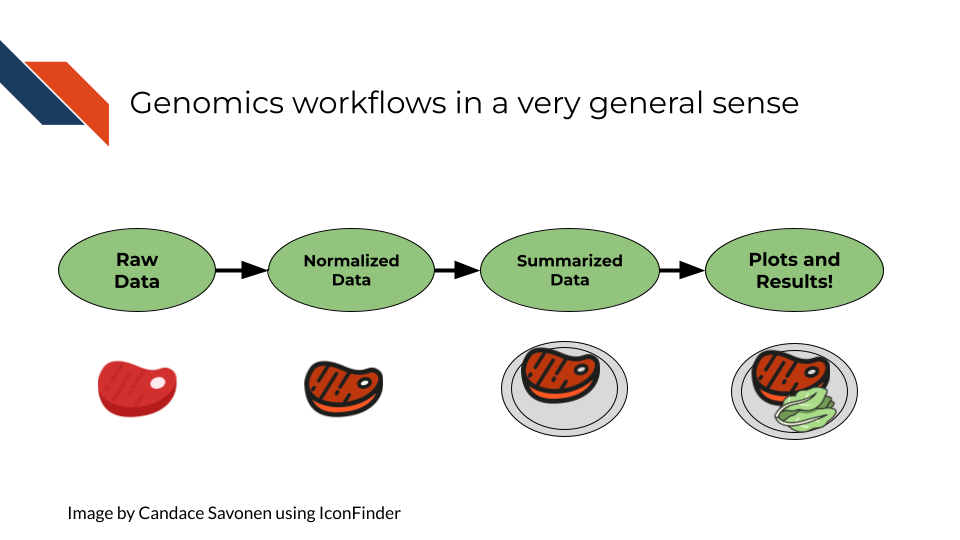
2.1.2 Basic file formats
Before we get into bioinformatic file types, we should establish some general file types that you likely have already worked with on your computer. These file types are used in all kinds of applications and not specific to bioinformatics.
2.1.2.2 TSV - Tab Separated Values
Tab separated values file is a text file is good for storing a data table. It has rows and columns where each value is separated by (you guessed it), tabs. Most commonly, if your genomics data has been provided to you in a TSV or CSV file, it has been processed and summarized! It will be your job to know how it was processed and summarized
Here the literal ⇥ represents tabs which often may show up invisible in your text editor’s preference settings.
gene_id⇥sample_1⇥sample_2
gene_a⇥12⇥15,
gene_b⇥13⇥142.1.2.3 CSV - Comma Separated Values
A comma separated values file is list just like a TSV file but instead of values being separated by tabs it is separated by… (you guessed it), commas!
In its raw form, a CSV file might look like our example below (but if you open it with a program for spreadsheets, like Excel or Googlesheets, it will look like a table)
gene_id, sample_1, sample_2,
gene_a, 12, 15,
gene_b, 13, 142.1.3 Sequencing file formats
2.1.3.1 SAM - Sequence Alignment Map
SAM Files are text based files that have sequence information. It generally has not been quantified or mapped. It is the reads in their raw form. For more about SAM files.
2.1.3.2 BAM - Binary Alignment Map
BAM files are like SAM files but are compressed (made to take up less space on your computer). This means if you double click on a BAM file to look at it, it will look jumbled and unintelligible. You will need to convert it to a SAM file if you want to see it yourself (but this isn’t necessary necessarily).
2.1.3.3 FASTA - “fast A”
Fasta files are sequence files that can be either nucleotide or amino acid sequences. They look something like this (the example below illustrating an amino acid sequence):
>SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT2.1.3.4 FASTQ - “Fast q”
A Fastq file is like a Fasta file except that it also contains information about the Quality of the read. By quality, we mean, how sure was the sequencing machine that the nucleotide or amino acid called was indeed called correctly?
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65Later in this course we will discuss the importance of examining the quality of your sequencing data and how to do that. If you received your data from a bioinformatics core it is possible that they’ve already done this quality analysis for you.
Sequencing data that is not of high enough quality should not be trusted! It may need to be re-run entirely or may need extra processing (trimming) in order to make it more trustworthy. We will discuss this more in later chapters.
2.1.3.5 BCL - binary base call (BCL) sequence file format
This type of sequence file is specific to Illumina data. In most cases, you will simply want to convert it to Fastq files for use with non-Illumina programs.
2.1.3.6 VCF - Variant Call Format
VCF files are further processed form of data than the sequence files we discussed above. VCF files are specially for storing only where a particular sample’s sequences differ or are variant from the reference genome or each other.
This will only be pertinent to you if you care about DNA variants. We will discuss this in the DNA seq chapter.
For more on VCF files.
2.1.3.7 MAF - Mutation Annotation Format
MAF files are aggregated versions of VCF files. So for a group of samples for which each has a VCF file, your entire group of samples’ variants will be summarized in the form of a MAF file.
For more on MAF files.
2.1.4 Microarray file formats
2.1.4.1 IDAT - intensity data file
This is an Illumina microarray specific file that contains the chip image intensity information for each location on the microarray. It is a binary file, which means it will not be readable by double clicking and attempting to open the file directly.
Currently, Illumina appears to suggest directly converting IDAT files into a GTC format. We advise looking into this package to help you do that.
2.1.4.2 DAT - data file
This is an Affymetrix’ microarray specific file parallel to the IDAT file in that it contains the image intensity information for each location on the microarray. It’s stored as pixels.
2.1.4.3 CEL
This is an Affymetrix microarray specific file that is made from a DAT file but translated into numeric values. It is not normalized yet but can be normalized into a CHP file.
2.2 General informatics files
At various points in your genomics workflows, you may need to use other types of files to help you annotate your data. We’ll also discuss some of these common files that you may encounter:
2.2.0.1 BED - Browser Extensible Data
A BED file is a text file that has coordinates to genomic regions. THe other columns that accompany the genomic coordinates are variable depending on the context. But every BED file contains the chrom, chromStart and chromEnd columns to start.
A BED file might look like this:
chrom chromStart chromEnd other_optional_columns
chr1 0 1000 good
chr2 100 3000 badFor more on BED files.
2.2.0.2 GFF/GTF General Feature Format/Gene Transfer Format
A GFF file is a tab delimited file that contains information about genomic features. These types of files are available from databases and what you can use to annotate your data.
You may see there are GFF2, GFF3, and GTF files. These only refer to different versions and variations. They generally have the same information. In general, GFF2 is being phased out so using GFF3 is generally a better bet unless the program or package you are using specifies it needs an older GFF2 version.
A GFF file may look like this (borrowed example from Ensembl):
1 transcribed_unprocessed_pseudogene gene 11869 14409 . + . gene_id "ENSG00000223972"; gene_name "DDX11L1"; gene_source "havana"; gene_biotype "transcribed_unprocessed_pseudogene";Note that it will be useful for annotating genes and what we know about them.
2.2.1 Other files
* If you didn’t see a file type listed you are looking for, take a look at this list by the GSEA developer team. Or, it may be covered in the data type specific chapters.