
Chapter 18 ChIP-Seq
This chapter is in a beta stage. If you wish to contribute, please go to this form or our GitHub page.

18.2 What are the goals of ChIP-Seq analysis?
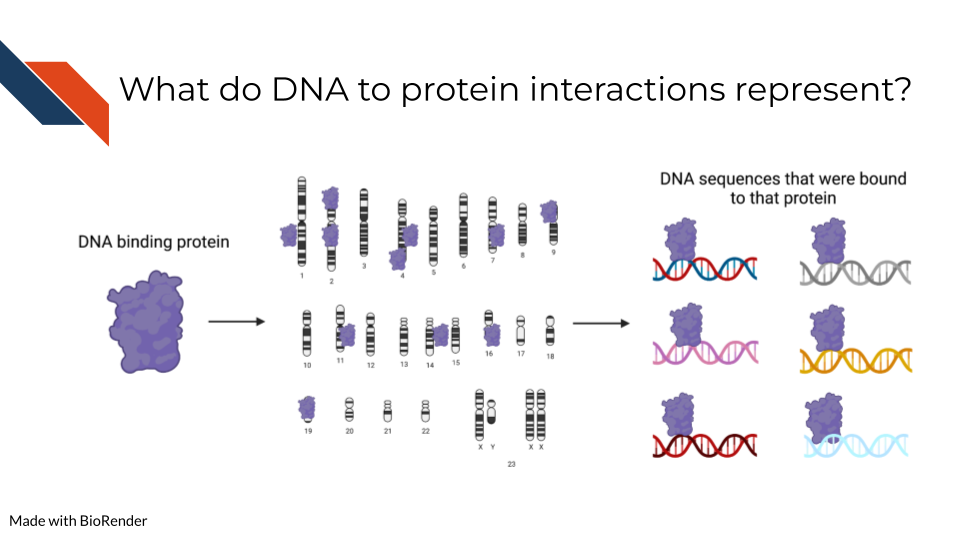
ChIP-Seq (chromatin immunoprecipitation sequencing) and related approaches are used to identify genome-wide binding sites of specific proteins or protein complexes. Given the diversity of interactions at the DNA-protein interface, sequencing-based methods for targeted chromatin capture have evolved to meet precise research needs and improve the quality of the results. Specifically, ChIP-Seq builds on protein immunoprecipitation techniques (IP) by applying next generation sequencing to a pulldown product. IP followed by sequencing can be applied to any nucleic-acid binding protein for which an antibody is available, including a known or putative transcription factor (TF), chromatin remodeler or histone modifications, or other DNA- or chromatin-specific factors. ChiP-Seq approaches have been honed to increase signal-to-noise, reduce input material, and more specifically map protein-DNA interactions, for example by treating the IP product with a exonuclease that chews-back unprotected DNA end (e.g. ChIP-exo).
The main goals of analysis for ChIP-Seq approaches are:
- Identify the genomic regions where a specific protein or protein complex binds. This can be achieved by sequencing both the IP input and product, and then calculating the enrichment in the product sample over the input.
- Annotate binding sites via comparison to other datasets and genome annotations. This may include transcription start sites (TSSs) or gene-regulatory regions. Oftentimes it is best to validate your data against previous profiling of similar epitopes.
- Comparison of binding sites: Many ChIP-Seq experiments compare changes in protein-DNA interactions across different conditions. This type of analysis can leverage statistical tools for pairwise comparison and multiple hypothesis testing.
- Identification of co-occurring motifs: Many chromatin proteins exhibit a sequence-specific binding pattern that is shaped by evolutionary forces. These sequence patterns, or motifs, are thought to capture contacts between specific base pairs and the DNA-binding domain of a protein and are often represented as a position weight matrix (PWM) for computational analysis. Statistical tools have been developed for de novo motif discovery within a given set of genomic intervals, like a ChIP-seq peaklist. The list of discovered motifs can be meaningfully interpreted by cross referencing with a motif database and recovery of known motifs represent another means of data validation.
- Integration with other -omics data: Given the expansive repositories of publicly available sequencing data, creating a comprehensive narrative from a ChIP-Seq experiment usually involves comparison with other types of sequencing data. Just like how a ChIP-Seq peak list can be interpreted through existing genome annotations, other sequencing data can be interpreted through the binding sites identified from a given ChIP-Seq experiment. For example, a sequence variant might be enriched for or against in protein binding sites versus previously identified motifs. This would suggest that a mutation would alter DNA-protein interactions. Binding of a specific gene-regulatory element might also correlate with changes in gene expression.
18.3 ChIP-Seq general workflow overview
A key contribution of large consortia, such as the ENCODE consortium, are standardized processing workflows to facilitate the integration of ChIP-seq data generated in different labs. While the exact data processing needs of any given experiment may vary, established pipelines provide a helpful starting point. In choosing a data processing workflow, it is essential to note the input data format. For example, the read length should be considered, as well as the sequencing paradigm (i.e. whether the data is single-end or paired-end).
The most generic steps for processing ChIP-Seq data are:
- Quality control: The first step in ChIP-Seq data processing is to perform quality control checks on the raw sequencing data to assess its quality and identify any potential issues, such as poor sequencing quality or adapter contamination which can be assessed via FASTQC.
- Read alignment: The next step is to align the ChIP-Seq reads to a reference genome using a suitable alignment tool such as Bowtie or BWA. Notably, many publicly available ChIP-Seq datasets are single-ended and it is important to use the correct alignment parameters for a given sequencing approach. In the case of ChIP-seq approaches that include exonuclease treatment, such as ChIP-exo and ChIP-nexus, a paired-end sequencing approach is often taken and then insert size can be useful for validating alignment. For example, profiling of a histone modification should yield nucleosome-sized fragments, ranging up from 120 bp for mononucleosomes, whereas TFs should yield smaller, sub-nucleosomal fragments and polymerase is in between at 20-50bps (PMID: 30030442).
- Peak calling: After the reads have been aligned to the genome, the next step is to identify the genomic regions where the protein or protein complex of interest is bound. This is done using peak-calling algorithms, such as MACS2, SICER, or HOMER, which can calculate enrichment as fold change over the input control with statistical testing.
- Quality control of peaks: Once the peaks have been called, it is important to perform quality control checks to ensure that the peaks are of high quality and biologically relevant. This can be done by assessing the number of peaks, fraction of reads in peaks (FRiPs), enrichment of the peaks in specific genomic regions, comparing the peaks to known gene annotations, or performing motif analysis. Often, peaks will be merged across replicates to create a consensus peak set. Peaks should be assessed visually with tools like IGV or the UCSC genome browser to ensure they overlap regions of high coverage. The Cistrome Data Browser is another useful resource for comparing with published ChIP-seq, DNase-seq and ATAC-seq data.
- Differential binding analysis: If the ChIP-Seq experiment involves comparing the binding of the protein or protein complex in different conditions or cell types, statistical testing can be performed to identify the regions of the genome where the protein or protein complex binds differentially. Tools developed for multiple comparison testing, like Limma, Deseq2, and EdgeR are useful for this type of comparative analysis.
- Integrative analysis: Finally, integrative analysis with other -omics data can be performed to gain biological insights into the ChIP-Seq data. This can involve interpreting ChiP-Seq data through existing annotations by looking at signal enrichment in different genomic regions, like transcription start sites (TSSs), gene bodies, and previously-identified cis-regulatory elements (CREs). ChIP-Seq data can even be interpreted through other ChIP-seq data to see if features overlap with statistical testing for similarity using packages like BEDTools and Bedops.
18.4 ChIP-Seq data strengths:
ChIP-Seq (chromatin immunoprecipitation sequencing) is a powerful tool for understanding the genomic locations where a specific protein or protein complex binds.
ChIP-Seq is particularly good at showing or illustrating:
- Identification of regulatory elements: ChIP-Seq can be used to identify the genomic regions where a protein or protein complex binds to regulatory elements, such as promoters, enhancers, and silencers. For example, certain histone modifications characterize active promoters and enhancers, such as H3K4 methylation and H3K27 acetylation.
- Characterization of protein-protein interactions: ChIP-Seq can be used to identify the genomic regions where multiple proteins bind. In this way, cobinding can be inferred to provide insight into the protein-protein interactions that are involved in regulating gene expression.
- Identification of binding site motifs: ChIP-Seq can be used to identify the DNA motifs that are enriched in the binding sites of a protein or protein complex. This information can be used to identify other transcription factors or cofactors that are involved in the same regulatory network. Databases of known TF binding motifs include JASPAR, Cis-BP, Hocomoco.
- Differential binding analysis: ChIP-Seq can be used to compare the binding of a protein or protein complex in different conditions or cell types, which can provide insight into the mechanisms that regulate protein binding and the impact of different cellular states on the regulatory networks.
18.5 ChIP-Seq data limitations:
ChIP-Seq (chromatin immunoprecipitation sequencing) is a powerful technique, but there are several biases, caveats, and problems that can arise when analyzing ChIP-Seq data.
Some of the most common biases, caveats, and problems are:
- Accessibility bias: ChIP-Seq relies on fragmentation of chromatin prior to immunoprecipitation, which is observed to enrich for genomic regions that are highly accessible to TFs in general .
- Antibody specificity and cross-reactivity: The specificity of the antibody used in ChIP-Seq is crucial for the accuracy of the results. Finding an antibody for specific epitopes can pose a challenge because antibodies can have cross-reactivity with other epitopes, which can result in false positives or misinterpretation of the data.
- DNA fragmentation bias: The length and quality of the DNA fragments used in ChIP-Seq can impact the results. Shorter fragments are often located in regions with more highly accessible chromatin, especially nucleosome linker regions and promoters of active genes. Sequencing depth bias: The amount of sequencing depth can impact the results of ChIP-Seq analysis. Insufficient sequencing depth can result in false negatives or miss important binding sites.
- Reproducibility and sample variation: ChIP-Seq experiments can be highly variable, and reproducibility between replicates can be an issue. Additionally, the composition and quality of the sample can also impact the results. Peak-calling algorithm choice: The choice of peak-calling algorithm can impact the results of ChIP-Seq analysis, as different algorithms have different strengths and weaknesses.
- Interpretation of binding sites: Finally, the interpretation of binding sites identified by ChIP-Seq can be complex and requires additional validation to confirm their biological relevance and function. Notably, ChIP-Seq cannot distinguish direct protein-DNA interaction from indirect binding (e.g. where a protein may bind another protein that binds to DNA).
18.6 ChIP-Seq data considerations
As a general guideline, a minimum sequencing depth of 20 million reads is recommended for ChIP-seq experiments in Drosophila, whereas 40–50 million reads is a practical minimum for most marks in human tissue (PMID: 24598259). However, this depth may not be sufficient for some analyses, particularly for studies that require high resolution or low signal-to-noise ratio. In such cases, deeper sequencing may be necessary to achieve the desired level of sensitivity and specificity.
In general, epitopes that cover large sequence space (e.g. repressive histone modification such as H3K27me3) require greater sequencing depth than epitopes confined to more narrow genomic regions (e.g. active histone modifications such as H3K4 methylation and H3K27ac). ChIP-seq for TFs may require even less sequencing depth; however, low antibody specificity may necessitate deeper sequencing due to low signal-to-noise. In practice, the depth of sequencing required for ChIP-seq experiments can vary widely depending on the specific experimental design and research question. It is important to perform a pilot study or use appropriate statistical methods to estimate the necessary sequencing depth for a given experiment. Choosing a specific antibody is essential, otherwise even deep sequencing may not recover signal over high background. Sequencing depth should also account for genome size (e.g. larger genome requires deeper sequencing).
18.7 ChiP-seq analysis tools
18.7.1 Tools for quality checks
- FastQC is a widely used tool that is used to assess the quality of sequencing data. It analyzes the raw sequencing data and generates a report that provides an overview of various metrics such as base quality, sequence length distribution, and GC content.
- Picard tools and SAMtools: Picard tools and SAMtools are two collections of command-line tools that are used to manipulate and analyze high-throughput sequencing data. They can be used to check the quality of the data, remove duplicates, and generate summary statistics.
- MACS2 (Model-based Analysis of ChIP-Seq) is a software tool that is specifically designed for the analysis of ChIP-Seq data. It is used to identify regions of the genome that are enriched for DNA-protein interactions.
- ENCODE Uniform Processing Pipelines: The ENCODE (Encyclopedia of DNA Elements) Uniform Processing Pipelines are a set of standardized protocols and tools that are used to process and analyze ChIP-Seq data. They ensure that the data generated by different labs are consistent and can be easily compared.
These tools are just a few examples of the many quality control tools available for ChIP-Seq analysis. The choice of tool(s) to use will depend on the specific analysis being performed and the preferences of the user.
18.7.2 Tools for Peak calling:
- MACS2 (Model-based Analysis of ChIP-Seq) is a widely used tool for peak calling in ChIP-Seq data. It uses a Poisson distribution to model the local noise and identifies peaks based on the fold enrichment over the background noise.
- SICER: Spatial Clustering for Identification of ChIP-Enriched Regions (SICER) is a peak caller that takes into account the spatial clustering of enriched regions in ChIP-Seq data. It uses a clustering algorithm to identify peaks based on the local density of enriched regions.
- HOMER (Hypergeometric Optimization of Motif EnRichment) is a suite of tools that includes a peak caller for ChIP-Seq data. It uses a sliding window approach to identify peaks based on the local enrichment of reads.
- PeakSeq is a peak caller that uses a Bayesian approach to identify enriched regions in ChIP-Seq data. It models the relationship between the read counts and the signal-to-noise ratio and identifies peaks based on the posterior probability of enrichment.
18.7.3 Tools for Differential Analysis
- DESeq2: This is a widely used R package for differential analysis of sequencing count data, including ChIP-seq. It uses a negative binomial model to normalize and test for differential enrichment of ChIP-seq peaks.
- edgeR: Another popular R package for differential expression analysis of RNA-seq data, edgeR can also be used for differential analysis of ChIP-seq data. It uses a generalized linear model to estimate differential enrichment and has been shown to be effective for ChIP-seq data with low read counts. Annotation
- ChIPseeker: This R package can be used for annotating ChIP-seq peaks with genomic features such as gene annotation, gene ontology, and pathway analysis. It can also generate plots and heatmaps for visualization.
- HOMER: This suite of tools includes several programs for motif discovery, peak annotation, and visualization. The annotatePeaks.pl program can be used for assigning genomic regions to specific functional categories, including promoter, exon, intron, intergenic, and enhancer regions.
- GREAT: This web-based tool can be used for annotating genomic regions with functional annotations such as gene ontology terms and regulatory domains. It uses a statistical approach to associate genomic regions with biological functions.
- Cistrome-GO: A web-based tool for determining the gene ontologies of genes likely to be regulated by regions discovered through TF ChIP-seq.
- GenomicRanges: This R package provides a framework for working with genomic ranges, including intersection, overlap, and annotation of genomic regions with functional categories. It can be used in conjunction with other R packages for ChIP-seq analysis, such as ChIPseeker and DiffBind.
- ChIP-Enrich: This web-based tool can be used for annotating ChIP-seq peaks with functional categories such as gene ontology, pathway analysis, and transcription factor binding sites. It uses a hypergeometric test to identify overrepresented functional categories.
- Cistrome DB: The website allows users to upload their enriched regions, returning TF ChIP-seq, DNase-seq or ATAC-seq samples with similar profiles.
18.7.4 Motif Analysis
- MEME Suite: The MEME Suite is a comprehensive suite of tools for motif analysis, including motif discovery and motif-based sequence analysis. It includes tools for discovering de novo motifs from ChIP-Seq data and for searching for known motifs in the regions bound by the protein of interest.
- HOMER is a suite of tools for motif discovery and analysis. It includes tools for identifying de novo motifs from ChIP-Seq data, as well as for searching for known motifs in the regions bound by the protein of interest. HOMER also provides tools for performing gene ontology analysis and pathway analysis based on the identified motifs.
- MEME-ChIP is a specialized version of the MEME Suite that is specifically designed for motif analysis in ChIP-Seq data. It includes tools for discovering de novo motifs from ChIP-Seq data, as well as for searching for known motifs in the regions bound by the protein of interest.
- CentriMois a tool for identifying enriched motifs in ChIP-Seq data based on the position of the motif relative to the peak summit. It can be used to identify motifs that are enriched at the center of the peak, as well as those that are enriched near the edges of the peak.
18.7.5 Tools for preprocessing
- Trimmomatic is a widely used tool for trimming and filtering Illumina sequencing data. It is often used to remove low-quality reads, adapter sequences, and other artifacts that can affect downstream analysis.
- Cutadapt is another popular tool for trimming adapter sequences from high-throughput sequencing data. It is particularly useful for removing adapters that contain degenerate nucleotides or that have been ligated with variable lengths.
- Bowtie2 is a fast and memory-efficient tool for aligning sequencing reads to a reference genome. It is often used to map ChIP-Seq reads to the genome prior to peak calling.
- SAMtools is a suite of tools for manipulating SAM/BAM files, which are commonly used to store alignment data from high-throughput sequencing experiments. It can be used for filtering and sorting reads, as well as for generating summary statistics.
- BEDTools is a powerful suite of tools for working with genomic intervals, such as those generated by ChIP-Seq peak calling. It can be used for operations such as intersecting, merging, and subtracting intervals.
18.7.6 Tools for making visualizations
- Integrative Genomics Viewer (IGV) is a popular genome browser that is widely used for the visualization of genomic data, including ChIP-Seq data. It provides a user-friendly interface for exploring genomic data at different levels of resolution, from the whole-genome level down to individual nucleotides.
- The UCSC Genome Browser is another widely used genome browser that can be used to visualize ChIP-Seq data. It provides an intuitive interface for navigating and visualizing genomic data, including the ability to zoom in and out and to overlay multiple data tracks.
- Genome Visualization Tool (GViz) is a package for the R statistical computing environment that provides functions for generating publication-quality visualizations of genomic data, including ChIP-Seq data. It offers a high degree of flexibility and customization, allowing users to create complex and informative plots that convey the relevant information in a clear and concise manner.
- UCSC Xena is a web-based visualization tool for multi-omic data and associated clinical and phenotypic annotations. It can be used with ChIP-seq data.
- Cistrome-Explorer A web-based visualization of compendia of ATAC-seq and histone modification ChIP-seq data for diverse samples, represented as a heatmap. Users can upload their ChIP-seq peak sets to assess the tissue specificity of their regions on the genome.
18.7.7 Tools for making heatmaps
- Deeptools is a widely used package for analyzing ChIP-seq data, and it includes a tool called “plotHeatmap” that can generate heatmaps from ChIP-seq data.
- Integrative Genomics Viewer (IGV) is a popular tool for visualizing and exploring genomic data. It includes a heatmap function that can be used to generate heatmaps from ChIP-seq data.
- EnrichedHeatmapis an R package for making heatmaps that visualize the enrichment of genomic signals on specific target regions.
- SeqMonk is a software package designed for the visualization and analysis of large-scale genomic data. It includes a heatmap function that can generate heatmaps from ChIP-seq data.
- ngs.plot is a tool that can generate different types of plots, including heatmaps, from NGS data. It includes a ChIP-seq specific mode that can be used to generate heatmaps from ChIP-seq data.
- ChAsE: ChAsE (ChIP-seq Analysis Engine) is a web-based platform for ChIP-seq analysis that includes a heatmap function that can generate heatmaps from ChIP-seq data.
These tools allow users to generate heatmaps of ChIP-seq data, which can be used to identify enriched regions of binding and to visualize patterns of binding across genomic regions.
The Cistrome Project has a large collection of human and mouse ChIP-seq, DNase-seq and ATAC-seq data, as well as tools for analyzing user generate ChIP-seq data with publicly available samples. These tools include the Cistrome Data Browser toolkit function that can find publicly available datasets that are similar to a ChIP-Seq peak set, and Cistrome-GO for gene ontology analysis of TF ChIP-seq target genes.
18.8 More resources about ChiP-seq data
- Shirley Liu’s Computational biology course
- Galaxy ChIP-seq tutorial
- ENCODE ChiP-seq tutorial
- Crazyhottommy’s ChIp-seq tutorial
- Harvard CUT&RUN tutorial
- 4DN CUT&RUN tutorial
- Henikoff Lab CUT&Tag tutorial
- ARCHS4 (All RNA-seq and ChIP-seq sample and signature search) is a resource that provides access to gene and transcript counts uniformly processed from all human and mouse RNA-seq experiments from GEO and SRA.
- UCSC Xena is a web-based visualization tool for multi-omic data and associated clinical and phenotypic annotations. It can be used with ChIP-seq data.
- Integrative Genomics Viewer (IGV) is a track-based browser for interactively exploring genomic data mapped to a reference genome.