graph LR A[Lockfile] --> B B[Binder Ready] --> C C[Dockerfile]
Making Code Ready for Publication
Reminder
This workshop adheres to the DaSL Learning Community Participation Guidelines:
Please be respectful of your fellow learners and help each other learn.
Remember, it’s dangerous to learn alone! So partner up with someone, it’s fun to learn together.
Introduction (you)
Introduce yourself live or in chat:
- Your name + pronouns
- Your group
- How do you use spreadsheets in your work?
- Favorite winter activity
Hit Record in Teams
TL; DR
- Reproducibility is a spectrum
- Use separate folders for each of your projects
- Organize code and Data
- Generate lockfiles
- Get Help from DaSL / Open Sci Organizations
- Deposit data where required by papers
Outline
- Why make your code ready for publication?
- What do you need to make it ready?
- How and where do you make it available?
Who this is for
- Majority of us want to share analyses, not software
- Leverage some principles from software packaging to share scripts and notebooks
- Software packaging is its own topic
“It worked on my machine”
- What are some issues that you’ve encountered in sharing your analyses with other people?
Why?
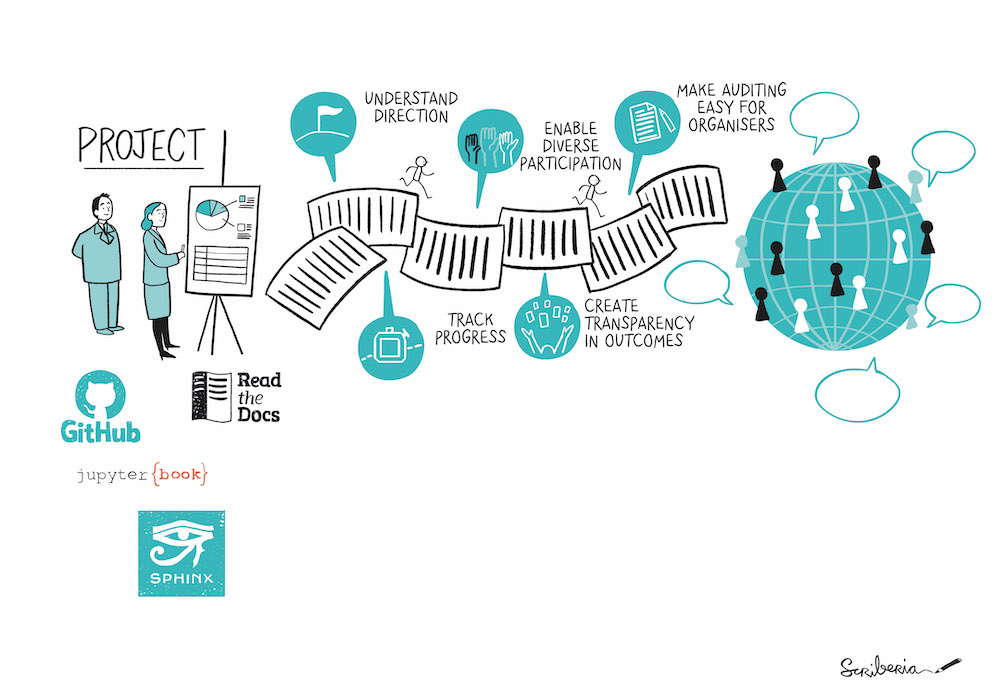
Why: Documentation is Important
- Do it for Future You
- Others in your lab
- Others in your field
Why: Reproducibility Matters
- Duke Medicine biomarker scandal
- Software is an important output
Why: Reproducibility is a Spectrum
- Do what you can
- Providing a good framework for running analyses
- Who is going to look at your code?
- Where are you going to share it?
Why: Reproducible Analyses are not perfect
- They only need to be able to run on another machine
- Don’t let perfect be the enemy of good
Why: Languages are Moving Targets
- Packages may depend on certain versions of Python/R
- Dependency Hell
- Need a way to “freeze” or “pin” versions used in analysis
- Language Versions
- Package Versions
Why: Reproducibility is an iterative process
- When possible, start from the beginning
- Use package management and environments from the start
rv/uv(in Package management session)
- Test out running scripts and notebooks as you go
What: Parts of a Reproducible Project
What: Minimum Information for Analyses
- Focus on Data Analysis in R / Python
- Organize your analysis in a folder and share in a repository
What: Separate Folders
- Ensures portability across platforms
- If there is repeated code from another project, considering packaging at that code
What: Project Example:
my_project/ ## Top level
├── data/ ## Data directory
│ └── my_data.vcf
├─- output/ ## Share output
└── 01_preprocessing.R ## Scripts in order
└── 02_deseq2_analysis.qmd
└── 03_visualization.ipynb
├── renv.lock ## R Packages
├── requirements.txt ## Python Packages
└── README.mdMore project examples
What: README
my_project/ ## Top level
└── README.md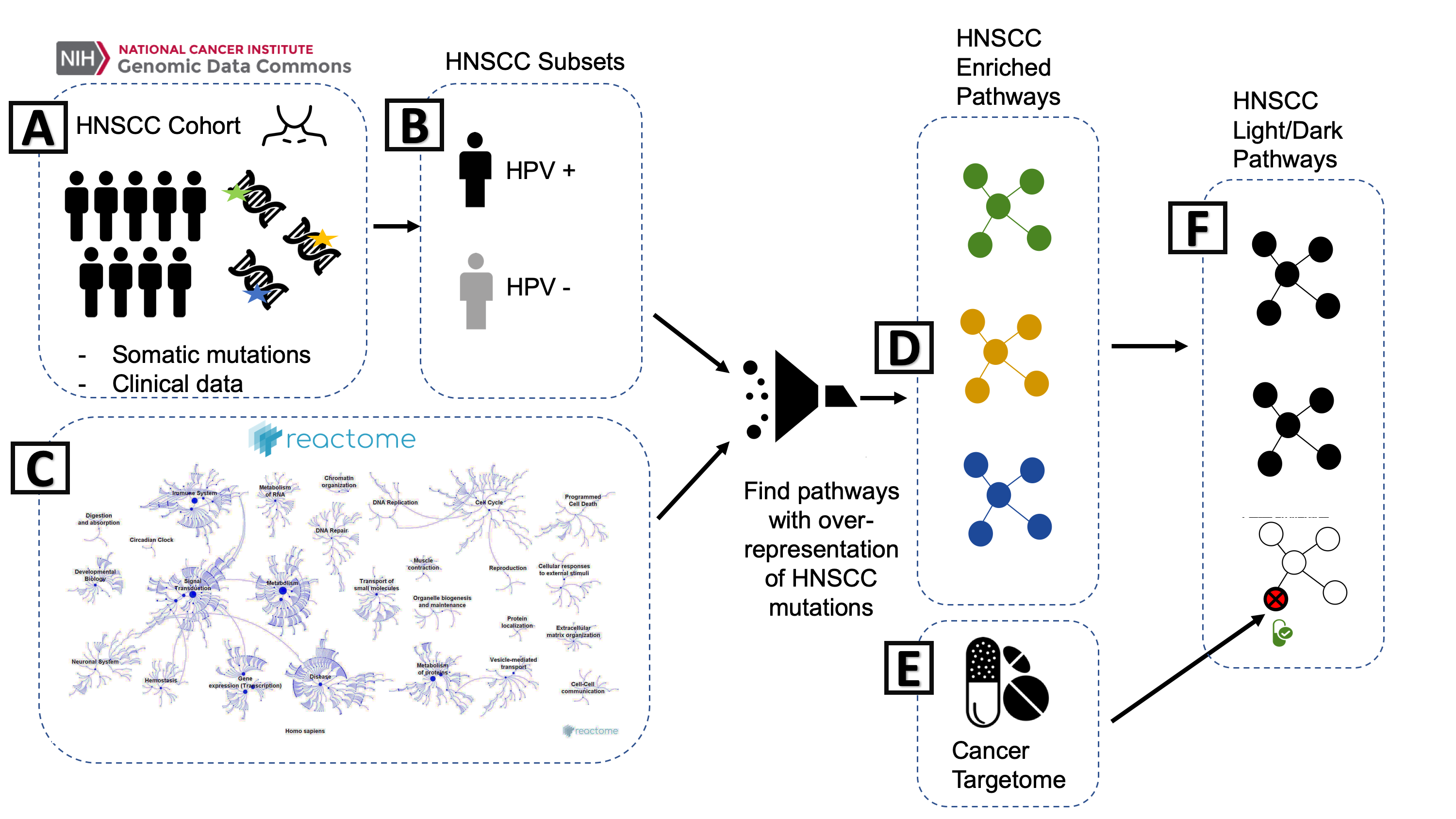
workflow.png
What: README
- First thing that people will see
- https://github.com/biodev/HNSCC_Notebook
- Document the basic workflow of processing
- How does the data come together in the analysis?
Exercise: Look at a README (5 min)
Pick one of these studies:
A. Integrative Pharmacogenomics Analysis of Patient Derived Xenografts (R)
B. BeatAML2 Manuscript Workflow (R)
C. An open RNA-Seq data analysis pipeline tutorial (Python)
Try and answer this question in the Google Doc
- How was the README? Was it Well Organized?
What: Notebooks / Analysis Files
my_project/ ## Top level
├── 01_preprocessing.R ## Scripts in order
├── 02_deseq2_analysis.qmd
└── 03_visualization.ipynb- Easiest: place in your top folder
- Number in order
01_preprocessing.R02_deseq2_analysis.qmd
- Be sure to include a random seed for reproducibility
What: Notebooks / Relative Paths
- Everything should be runnable from the top folder of the project. Put data in
data/folder. Use relative paths from the top project folder:
- Ensures portability of project
What: {targets} and Workflow Builders
- Not 100% Necessary!
- Another layer of complexity, but can be helpful
- Lets you chain together notebooks
- A lot like snakemake
Targets example: https://github.com/biodev/hnscc_manuscript
Exercise: Look at How Notebooks are Organized (5 min)
Pick one of these studies:
A. Integrative Pharmacogenomics Analysis of Patient Derived Xenografts (R)
B. BeatAML2 Manuscript Workflow (R)
C. An open RNA-Seq data analysis pipeline tutorial (Python)
Try and answer this question in the Google Doc
- How are the Notebooks organized?
What: Know the status of your data
- Is your data restricted or regulated for sharing?
- Schedule a Data Governance House Call if unsure
What: Data in a Project
my_project/ ## Top level
├── data ## Data directory
│ └── my_data.vcf - Genomic and omics data is large
- Raw data is not practical for GitHub (100 Mb limit)
- Store raw files in required respositories for your field
- Track what files were processed (manifest)
- Supply intermediate formats used to do the analysis, if possible:
- MAF/VCF/CSV
What: Data
With code, share metadata - list the files you processed
- File manifest - point towards data repositories
- JSON files from workflows
- Metadata / Experimental Design
- Where does each sample fit into Experimental Design?
Stay tuned - we may offer a data management workshop this Summer
Exercise: Examine how data is stored (5 minutes)
Pick one of these studies:
A. Integrative Pharmacogenomics Analysis of Patient Derived Xenografts (R)
B. BeatAML2 Manuscript Workflow (R)
C. An open RNA-Seq data analysis pipeline tutorial (Python)
Try and answer this question in the Google Doc
- How is the data stored (or not stored)?
What: Reproducible Environment
A Reproducible Environment is a computational environment is the system where a program is run.
- Operating System/Platform
- Language Version
- Package Versions
- Software Dependencies
What: Reproducible Environments
In order of complexity:
There is a tradeoff between - Effort on your side (Lockfile is least effort) - Ease of Use on User End (Dockerfile is most effort)
What: Lockfiles and Environments
- Lockfiles are a recipe for recreating an environment
- We use tools to recreate them:
- R:
renvandrv - Python:
venvanduv
- R:
- Environments live in our folder isolated from everything else
- Versions of R/Python
- Libraries of packages
What: Lockfile
my_project/ ## Top level
├── renv.lock ## R
├── requirements.txt ## Python- List of packages and versions that you used in analysis
- Talk about
rvanduvin Package management session
Lockfile Examples
{
"R": {
"Version": "4.2.3",
"Repositories": [
{
"Name": "CRAN",
"URL": "https://cloud.r-project.org"
}
]
},
"Packages": {
"markdown": {
"Package": "markdown",
"Version": "1.0",
"Source": "Repository",
"Repository": "CRAN",
"Hash": "4584a57f565dd7987d59dda3a02cfb41"
},
"mime": {
"Package": "mime",
"Version": "0.7",
"Source": "Repository",
"Repository": "CRAN",
"Hash": "908d95ccbfd1dd274073ef07a7c93934"
}
}
}Exercise: Look at a lockfile (5 minutes)
- R: https://github.com/fhdsl/mcr_example_r - look at
renv.lock - Python: https://github.com/fhdsl/mcr_example_r - look at
requirements.txt
Why not Conda?
Anaconda is charging institutions for using their forge - be aware that you will need to pay charges or change your forge to the Fred Hutch version.
For more info: https://conda-forge.fredhutch.org/
Exercise: Examine Reproducible Environments (5 minutes)
Pick one of these studies:
A. Integrative Pharmacogenomics Analysis of Patient Derived Xenografts (R)
B. BeatAML2 Manuscript Workflow (R)
C. An open RNA-Seq data analysis pipeline tutorial (Python)
Try and answer this question in the Google Doc
- How did they reproduce the software environment, or did they?
What: Reproducing environment from lockfile
- Download folder from GitHub
- Install language version (such as
R 3.4.3orPython 3.13) to your machine
What: Making a Lockfile from your current project
- Usually done after you execute a notebook
- Takes the packages you have loaded into memory and then writes them to lockfile with versions.
How
How and Where: Testing your shared project
- Try downloading and installing on a different computer to make sure that you can rerun analyses
- Take someone else through the process and test out the notebooks
- If making binder ready: test the repository on Binder
How: Review Opportunities and Resources
- Review Opportunities
- Data House Calls
- Community Co-working
- WILDS
- PyOpenSci and ROpenSci for code review if you decide to package your code
How and Where: Sharing Your Analyses
- GitHub (for code)
- Open Science Framework (for code + data)
- Field specific databases
- Social Media: LinkedIn, Bluesky, etc.
Where should you share code?
Share in a public repository:
- GitHub
- Codeocean
- ReadtheDocs
Be aware of file size limitations!
How: Data Repositories
- Data repositories
- Open Science Framework
- Required databases (dbGAP)
- Be aware that you will need to provide metadata
- Experimental design
- Be careful when sharing human subjects data
- If unsure, schedule a Data Governance House Call
https://journals.plos.org/plosgenetics/s/recommended-repositories#loc-omics
TL; DR
- Reproducibility is a spectrum
- Use separate folders for each of your projects
- Organize code and Data
- Generate lockfiles
- Get Help from DaSL / Open Sci Organizations
- Deposit data where required by papers
What’s Next?
- March 2: Package Management for R and Python
- March 11: Data Deep Dive on Workflows
- Next Quarter: Bash for Bioinformatics
- Join our Email list: send a blank email to dasltraining-join@lists.fhcrc.org
References
- https://book.the-turing-way.org/reproducible-research/compendia/
- https://github.com/leipzig/awesome-reproducible-research
- https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1005510
- https://openscapes.github.io/series/core-lessons/coding-strategies.html
- https://hutchdatascience.org/Tools_for_Reproducible_Workflows_in_R/index.html
- https://www.nature.com/articles/s41467-021-25974-w
- https://journals.plos.org/plosone/s/materials-software-and-code-sharing - Materials, Software and Code
- https://osf.io/cb7z8/files/numa5 - AGILE Reproducible Paper Guidelines
- https://binderhub.readthedocs.io/en/latest/overview.html
- https://book.the-turing-way.org/reproducible-research/code-reuse
Advanced Topics
What: Binder Ready Repository
A special way to share your analysis
- Put your project on GitHub
- Can plug your repository link into
mybinder.org - Generates JupyterLab / RStudio / Shiny Server instance
What: Binder.org
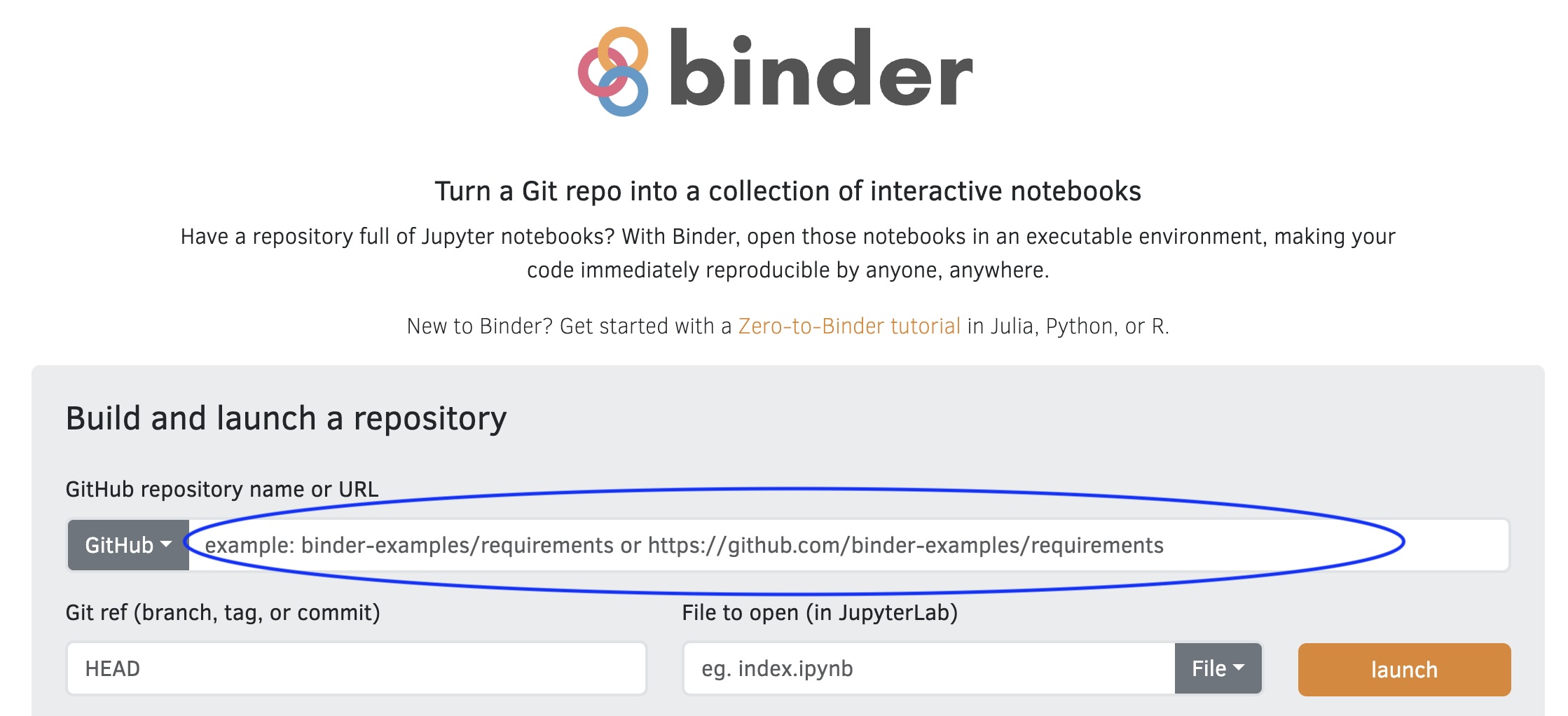
How does Binder work?
- Launches your analysis in a container (Dockerizes your analysis)
- Uses
requirements.txt(Python),environment.yml(Conda) orinstall.R(R) or Dockerfiles in your repository - Installs relevant packages and dependencies into a docker image
install.R using renv (put in your top directory)
Exercise: Launch a Binder Repository
- Go to mybinder.org
- Put in
Some Cons about Binder
- Currently limited to 2 Gb of memory for an instance
- If your image files aren’t used at least once a week, they get deleted
Dockerfile
- A precise list of instructions to install your computational environment
- More useful if you are distributing software.
- More portable across systems other than Binder
- Takes a lot of work, builds on the work of others
Dockerfile example
FROM debian:bookworm-slim AS builder
RUN Rscript
There will be a lot of crying
- Dockerfiles are a lot of work
Dockerfile Tips
- Don’t try to create Dockerfiles from scratch
- Community Images: BioC, Rocker Project, WILDS Docker Library
- Use https://repo2docker.readthedocs.io/en/latest/
- https://repo2docker.readthedocs.io/en/latest/configuration/#config-files
Nix
- Currently investigating Nix as a language agnostic reproducibility framework